Introduction
The biopharmaceutical industry (including small molecule drugs, biologics, and vaccines) and the medical equipment industry (including implantable medical devices, diagnostic imaging, and other diagnostics) have been major contributors to both rising healthcare spending and improved quality and quantity of life globally over the past four decades. Global spending on biopharmaceuticals reached one trillion dollars in 2012. Biopharmaceuticals account for between 10% and 20% of healthcare spending in most Organization for Economic Cooperation and Development countries, and often a higher share in developing countries that spend relatively less on hospital and physician services. The medical equipment sector is both conceptually less precisely defined and empirically harder to measure. Industry revenues are estimated at $332 billion (Ernst and Young, 2012), or roughly one-third of biopharmaceutical industry revenues.
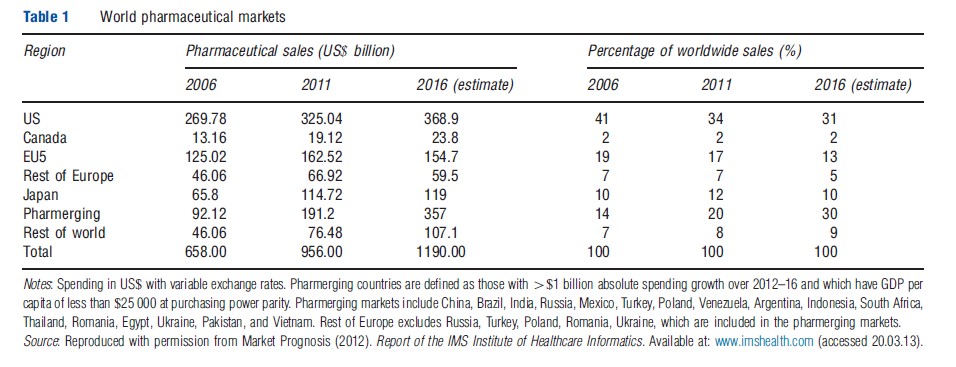
The US remains by far the largest single market for these industries. For biopharmaceuticals, the US share of global sales was 34% in 2011, down from 45% in 2000 (Table 1). Over the past decade growth of biopharmaceutical sales has slowed to low, single-digit annual growth rates in North America and Europe, due to patent expiries and genericization of many major drugs and slower growth of new drugs. This contrasts with double-digit growth of biopharmaceutical spending in many emerging markets, particularly China, Brazil, India, and some other countries of Asia, Africa, and Latin America, reflecting their rising incomes and increased spending on health care. For medical equipment, the US share is roughly 45% of global sales.
The economics literature has focused much more heavily on biopharmaceuticals than on medical devices and diagnostics, reflecting both the greater expenditure share of biopharmaceuticals and the greater availability of data. Economic analysis focuses on features that differentiate these industries from other health services or consumer goods industries, in particular: high research and development (R&D) intensity; heavy regulation of all business functions, including R&D, market access, pricing and marketing; and complex market environments due to physicians and payers being major customers, in addition to patients. Economic analysis has taken both a social welfare/policy perspective and a firm or industry perspective. From the policy perspective, key issues related to biopharmaceuticals are the design of intellectual property (IP) rights, regulatory and reimbursement systems to provide appropriate incentives for R&D, and to assure appropriate utilization and prices for drugs, devices, and diagnostics, such that they deliver value for money. From the firm or industry perspective, key issues include understanding the causes of declining R&D productivity and optimal strategic responses; measurement and demonstration of incremental value of new compounds to regulators and payers; and development of effective entry and sales strategies for emerging markets. Because regulation of market access, pricing, and reimbursement are decided by each country separately, global policy and strategy must consider the interaction of policies adopted in different countries, in particular, the many challenges related to segmentation and differential pricing when selling global products in markets that differ vastly in regulation, IP, and ability and willingness to pay.
This overview article on the economics of these industries lays out the theoretical issues and major empirical findings, focusing first on issues related to R&D and then turning to markets, reimbursement and pricing, promotion, and specific issues related to vaccines, personalized medicine, and biosimilars. Although this article focuses on biopharmaceuticals, reflecting the much larger literature, it also describes ways in which medical equipment is similar and different.
R&D: Costs, Regulation, And IP
R&D Costs And Regulation
The biopharmaceutical industry is unusually research intensive. The US research-based industry invests approximately 15% of its sales in R&D, compared with approximately 4% for US industry in general and 8% for the US-based medical device industry. The R&D cost of bringing a new medical entity (NME) to market is currently estimated to be approximately $1.5 billion (Mestre-Ferrandiz et al., 2012) and take 5–12 years from discovery through development, clinical trials, and regulatory approval. New drugs must meet stringent standards of safety, efficacy, and manufacturing quality before receiving market access approval. Large and lengthy clinical trials to demonstrate safety and efficacy, with high failure rates, are major drivers of the high cost per approved NME. Throughout the 1970s, 1980s, and 1990s, the cost per approved new drug increased by seven to eight percentage points per year above general price inflation. Factors contributing to rising cost per NME include not only rising clinical trial costs but also, more recently, higher failure rates. The evidence suggests that of drugs entering human clinical trials, only one in seven or eight reaches approval, compared to one in five in the 1990s. Rising failure rates reflect both safety, efficacy, and economic factors. Recent scientific advances have enabled development of novel therapies, but predictability remains imperfect. Further, because good treatments already exist for easier diseases, new drugs must now either provide significant incremental value relative to existing drugs that are available as low-priced generics, or tackle diseases that pose tougher scientific challenges, such as Alzheimer’s disease and cancer, or target diseases that were previously ignored due to small populations. Most recently approved drugs target either specialty conditions (complex, relatively uncommon diseases treated by specialists) or even small orphan indications (defined in the US as affecting less than 200 000 patients per year). In the US in 2010 and 2011, one-third of new active substances approved had orphan designation. This reflects the intended incentives provided by the Orphan Drug Act, which provides special tax credits and market exclusivities for drugs that receive orphan status, as well as the very high prices realized by some orphan drugs, now more than $400 000 per patient per year for some drugs. It also reflects the granting of orphan status for small indications for drugs that may subsequently be approved for other, larger indications – for example, many cancer drugs serve both orphan and nonorphan indications.
The cost of developing a new drug includes the out-of-pocket expenses incurred by firms from discovery through first approval on the successful compound and related failures, because failures are an unavoidable part of the process. The full, capitalized cost per approved NME also includes the opportunity cost of capital invested, because investors must recoup their opportunity cost in order to continue investing in R&D. This cost of capital is about half the total cost (Di Masi and Grabowski, 2007). Although the mean cost is estimated at US$1.5 billion (Mestre-Ferrandiz et al., 2012), there is significant variation with lower costs for rare diseases that necessarily have smaller trials, and relatively high costs for drugs to treat high-volume, chronic diseases that require large and long trials.
R&D expense for medical devices is much lower than that for drugs. Devices are classified into classes I through III, based on risk to patients and device novelty. The US Food and Drug Administration (FDA) has oversight over device safety, efficacy, and quality, but clinical trials are usually required only for novel devices classified as class III. Most devices are incremental modifications of existing products and can be approved by showing ‘substantial similarity’ to an existing device, without clinical trials. The EU’s CE mark system authorizes either state or private oversight bodies to review safety and quality, and proof of efficacy is not required. Devices are therefore often launched earlier in the EU than the US, in contrast to drugs for which EU launch is often delayed by reimbursement requirements.
Safety: Benefits And Costs
Market access regulation that requires demonstration of safety and efficacy entails costs as well as benefits. The appropriate extent and structure of this regulation has been debated in the academic and policy literatures. The main economic focus has been whether the current regulatory approach to drug approval provides an optimal trade-off between safety and delay. The benefits of regulation include preventing unsafe and ineffective drugs from being sold and requiring the production of unbiased information about drug outcomes, including risks, benefits, and contraindications as demonstrated in controlled trials. The statistically significant findings from clinical trials form the basis for the product label and approved promotional messages. By revealing the true expected benefits and risks from drugs before launch, such information reduces the risk of adverse outcomes and drug withdrawals for safety reasons.
The costs of market access regulation include increased development costs, which may keep some potential drugs off the market, and delay in consumer access to new drugs. The FDA User Fees (which fund the hiring of additional reviewers) and the Fast Track and Priority Review regulatory initiatives have accelerated the review process of new drugs and provided mechanisms for approval based on surrogate endpoints, with postlaunch follow-up. Despite some mixed evidence that more rapid reviews have resulted in more postlaunch adverse events and drug withdrawals, on balance the evidence from pharmaceuticals suggests that these initiatives have increased consumer welfare. For medical devices, the appropriate structure and requirements for review are still under debate in the US. Delays in approval relative to the EU are a concern, but so is the number of recalls of devices approved through the accelerated process. Future economic research is needed on the optimal structure of market access regulation for medical devices.
Patents, Exclusivities, And Other Research And Development Incentives
The high cost of R&D for biopharmaceuticals (and, to a lesser extent, medical devices) implies a cost structure with high fixed costs that can benefit consumers globally but are sunk at launch, with low marginal cost per pill. Investment in the costly and risky process of pharmaceutical R&D therefore requires some mechanism to assure a return on successful investments for originator firms. The standard approach is patents which grant the innovator a monopoly for the duration of the patent by barring identical copies. Defining appropriate patent terms and criteria for postpatent generic entry are critical policy issues. All countries that are members of the World Trade Organization must recognize 20-year product patents, running from date of filing, for all products that meet requirements of novelty and utility, not just pharmaceuticals.
In addition to this basic patent protection that applies to all types of goods, the US and many other countries have added regulatory provisions that define certain exclusivity protections for qualifying originator pharmaceuticals, partially make-up for patent term lost before launch due to the lengthy R&D process, and also define entry conditions for generics. In the US, the 1984 Hatch–Waxman Patent Restoration and Generic Competition Act extended patent terms and defined regulatory exclusivities for originators, and eased entry requirements for generic versions of small molecule drugs. Specifically, Hatch–Waxman provided originator drugs with up to 5 years of patent restoration to compensate for patent life lost during R&D and regulatory review, and 5 years of exclusivity for originator data before generics can Bibliography: the data. For generics, Hatch–Waxman provided an Abbreviated New Drug Approval (ANDA) pathway that enables generics to be approved without doing new safety and efficacy trials, provided they can show bioequivalence to the originator drug and Bibliography: the originator safety and efficacy data. Paragraph IV provides a 180-day market exclusivity for the first ANDA generic that successfully challenges originator patents, to incentivize challenge to dubious patents.
The ANDA provisions greatly reduced the regulatory costs of approval for generics and facilitated the growth of generics in the US. The 180-day exclusivity period has led to successful challenges of many patents, and hence speeded generic entry. Generics now account for more than 80% of all prescriptions dispensed in the US, and a higher percentage for compounds for which generics are available. Unsurprisingly, because patentability requires that an invention be new, useful, and nonobvious, original composition-of-matter patents that apply to new molecules have generally withstood generic challenge in the US, whereas additional patents filed later on ancillary features or new delivery systems have more frequently been successfully challenged for failing to meet requirements of novelty and nonobviousness. The requirements for proof of novelty and nonobviousness differ across countries. This has led to some products that are patented in the US being denied patents in countries such as India.
Given the experience of patent litigation and uncertainty under the Hatch–Waxman Act, the 2010 Affordable Care Act (ACA) provisions for a new regulatory approval pathway for follow-on biologics (biosimilars) has focused on the regulatory exclusivity period for originator data. This is currently set at 12 years from the first licensing of the Bibliography:d biologic, in contrast to 5-year data exclusivity for chemical drugs in the US. Whether this much longer exclusivity period, combined with more favorable reimbursement for biologics, potentially distorts R&D choices toward biologics, despite their lower convenience and higher cost for consumers, is an important topic for future research. In contrast to these discrepant US data exclusivity periods, the EU grants 10 years of data exclusivity for both chemical and biologic drugs.
More generally, regulatory exclusivities offer more flexibility of duration and more certainty of enforcement, compared to patents that must run for 20 years from filing but may be challenged. However, this flexibility may make regulatory exclusivities more subject to manipulation by special interests. Given the vastly different costs involved in different types of biopharmaceutical and medical technology R&D, use of both patents and the more flexible exclusivities seems optimal.
For medical devices, patents are important but in general create weaker and less durable market power than for pharmaceuticals, because it is relatively easy to invent around a medical device patent using a slightly different product design. Moreover, entry of incrementally improved, follow-on devices renders the original design obsolete within a few years, even if the 20-year patent nominally remains valid.
Although patents are in some respects an efficient and effective mechanism to incentivize R&D, patents have other disadvantages besides the inflexible term and uncertain validity already mentioned. In particular, patents operate by limiting competition and enabling innovator firms to charge prices above marginal cost, which can lead to suboptimal use of drugs in the absence of insurance. High price–marginal cost margins also create strong incentives for promotion. Several alternatives to patents have been proposed for pharmaceuticals, including both ‘push’ programs that provide subsidies to reduce the cost of R&D and ‘pull’ programs that increase and/or guarantee revenues for companies that bring new drugs to market, including prizes, patent buyouts, and advance market commitments. Some of these alternatives have been applied to R&D for ‘neglected’ diseases with prevalence predominantly in low-income countries, including the advance market commitment for the pneumococcal vaccine.
Further research is needed on the optimal mix of IP alternatives, including patents, exclusivities, and others, for specific R&D contexts related to drugs, devices, and other technologies, in order to appropriately reward innovation without granting inefficient barriers to entry. Such research should consider how the optimal mix of protections might differ across countries at different levels of development. Because the goal of IP or other protections is to provide an appropriate financial reward to innovators, the optimal type and duration of IP should ideally also consider the pricing and reimbursement environment, which determines the prices and revenues that can be earned during the protection period. More on this below.
Mergers, Alliances, And Organization Of R&D
The basic and translational science underlying many new drugs is developed in academic institutions, often supported by government research grants. The traditional mechanism for developing and commercializing such technologies has been the creation of start-up companies, usually with venture capital funding, taking advantage of the Bayh–Dole Act that encourages private commercialization of publicly funded research. Over the past two decades, thousands of start-up firms have been formed, many have been acquired by larger, established firms, some have failed, and a few have grown to become fully integrated biotechnology companies. Over time, the share of new approved drugs that originated with small firms has grown.
As large pharmaceutical firms have experienced declining returns on their internal R&D, they are increasingly using product licensing alliances and outright acquisition of small firms to source new compounds externally. For the small firms, such alliances with established biopharmaceutical firms provide an important source of R&D financing, as well as regulatory and commercial experience and expertise. The terms of these alliances and acquisitions are structured to align incentives and share risk, through payments that are triggered only if the product achieves certain goals. These contingent payments include R&D milestone payments, tiered sales royalties, opt-in options for the licensee in alliances, and contingent valuation rights linked to sales in acquisitions.
The theoretical literature has hypothesized that formation of product development alliances may be hampered by asymmetric information. However, contingent payments in the deal structure are designed to address both adverse selection and moral hazard risks. The empirical literature is mixed, but in general finds that in-licensed products have a higher probability of success than internally developed products, which supports the notion that the stringent due diligence process of alliance formation is more rigorous at weeding out compounds that will ultimately fail, compared to internal R&D review processes within large firms.
In addition to alliances with small firms, several large firms have recently reorganized their drug discovery divisions into small units that attempt to mimic the entrepreneurial spirit and incentives of small firms. The compounds that are produced by these internal units must compete with externally sourced compounds for scarce resources to fund clinical trials. Other attempts to increase R&D productivity within large firms include changes in personnel and organizational structure, and changes in compensation schemes. Despite all these attempts to improve R&D productivity, several large pharmaceutical companies have cut their R&D budgets recently for the first time in decades and instituted share buy-back programs, in response to shareholder concerns about the low return on R&D investment.
Small firms are not immune to the rising costs of R&D and high failure rates. Longer and riskier investment cycles and uncertainty of exit through either acquisition or an initial public offering have also slowed the flow of venture capital into formation of early-stage biotechnology companies. This decline in private equity and venture funding for start-ups has been partially offset by an increase in alliances directly between large pharmaceutical firms and academic institutions and a growth in funding through the corporate venture capital arms of large biopharma firms. These and other creative financing developments suggest that there may be efficiency gains from facilitating mechanisms to finance the development of new products without the formation of new start-up companies around each idea.
Markets For Biopharmaceuticals And Medical Technology
Principles Of Optimal Insurance
The market for pharmaceuticals in any country depends on the extent of insurance and on the rules of reimbursement used by payers to control the effects of insurance on prices and utilization. Insurance protects consumers against the financial risk of high drug spending but also makes consumers insensitive to drug prices. Demand-side price sensitivity is further undermined by the fact that physicians who prescribe drugs often lack the information and incentives to make pricesensitive choices. Inelastic demand of insured consumers creates incentives for firms to charge higher prices than they would if consumers were informed decision-makers facing full prices. To address this insurance-induced price insensitivity, insurers in most countries use a range of strategies to control prices and utilization of prescription drugs.
The optimal design of insurance coverage is a critical policy issue that affects patients’ access and financial exposure, innovation incentives for firms, and budget impact for taxpayers and consumers. In theory, insurance coverage and eligibility should be designed to encourage optimal utilization of existing drugs (static efficiency) and optimal incentives for R&D investment for new drugs (dynamic efficiency) and provide reasonable financial protection for patients. One proposed approach to achieving these three goals is that copayments should be set at marginal cost while the health insurer pays a top-up payment to the biopharmaceutical firm to reward innovation (Lackdawalla and Sood, 2009). In practice, both marginal cost and appropriate top-up payments are difficult to observe, and this approach ignores appropriate financial protection for patients.
An alternative approach, that could in theory achieve second-best static and dynamic efficiency and appropriate financial protection for patients, is for each payer to make reimbursement of a drug conditional on meeting an incremental cost-effectiveness ratio (ICER) threshold – for example, $50 000 per quality-adjusted life-year (QALY) – that reflects the willingness-to-pay for health gain of that payer’s enrollees or citizens (Danzon et al. 2012). The firm would be permitted to price up to the ICER threshold, but this implies that the price premium would be constrained by the new drug’s incremental benefit relative to the comparator or standard of care. The payer would also define coverage eligibility to assure access for patients for whom the drug is cost-effective at the price charged. Copayments would be modest, to collect some revenue but assure affordability. This approach encourages appropriate innovation, by paying a premium for new drugs that is based on their incremental value, and assures access for patients. If all countries with comprehensive insurance set ICER thresholds unilaterally, based on their willingness to pay for health, manufacturers would have incentives to set prices that differ across countries, reflecting countries’ willingness and ability to pay. This result is broadly consistent with Ramsey pricing principles applied to R&D as a joint cost.
In practice, pharmaceutical pricing and reimbursement regulation differs across countries but follows four broad prototypes: (1) the USA exemplifies free pricing in a pluralistic insurance market with competing health plans; (2) Europe exemplifies several approaches to setting price and reimbursement in universal insurance systems; (3) Japan exemplifies price regulation in a market where physicians traditionally dispensed drugs; and (4) many emerging markets illustrate predominantly self-pay markets for drugs. The following sections describe key economic issues in each of these prototypical markets.
Free Pricing With Competing Payers: The US
In the pluralistic US healthcare system, no single payer has sufficient market power to significantly influence prices. Payers rely primarily on tiered formularies and costs-haring to preserve some patient price-sensitivity and to enable payers to negotiate discounts in return for preferred formulary status. Although list prices are unconstrained, tiered formularies have achieved significant discounts in therapeutic classes with close therapeutic substitutes. However, in classes with few and/or differentiated products, which includes most specialty drugs and biologics, payers have not used tiered formularies aggressively to attempt to extract discounts. Rather, they rely increasingly on specialty tiers with 20–30% coinsurance rates. However, most patients are protected by catastrophic limits on costs-haring or manufacturer copay coupons, which provides appropriate financial protection but leaves little if any constraint on prices. Launch prices for new drugs therefore continue to rise, with several more than $100 000 per year or per treatment course. Similarly, for physician-dispensed biologics, the reimbursement rules create incentives for high launch prices, with little constraint from patient costs-haring.
By contrast, generic markets in the US are highly price competitive. High rates of generic entry and penetration, combined with low generic prices, reflect not only the Hatch–Waxman provisions requiring bioequivalence with low entry costs, but also pharmacy substitution and reimbursement rules that assure price-conscious dispensing choices by pharmacies and patient acceptance of generics. Over the past 15 years, patent expiration on many originator drugs has enabled a massive shift toward generics. In 2012, more than 80% of prescriptions were dispensed generically, up from 47% in 2000, but generics account for only approximately 30% of sales by value, due to their low prices. Generic penetration rates are higher and generic prices are absolutely lower in the US than in many other countries (Danzon and Furukawa, 2011). This has provided significant savings to consumers and created budget headroom for high-priced new drugs. As the flow of new generics declines, attention may shift to better ways to assure value for money while preserving access to new pharmaceuticals in the US.
Effects Of Cost Sharing
Patient cost sharing is an important feature of health-insurance design, particularly in the US. In theory, optimal cost sharing balances financial protection of patients against deterring overuse of services and excessive pricing. If other constraints on pricing or use are also used, then optimal cost sharing can be lower. Conversely, Garber et al. (2006) show that at levels of cost sharing that are optimal for patient protection, prices would exceed levels needed to incentivize optimal R&D, assuming current patent design is optimal. Unsurprisingly, cost-sharing levels are highest and studies of cost-sharing effects are most numerous in the US.
Because details of cost-sharing structure, levels, stop-loss, and other controls differ across contexts, generalizations are problematic. With that caveat, the evidence confirms that tiered cost sharing affects choices between drugs. Even modest cost sharing affects utilization and compliance. Recent studies have focused on the interconnection between utilization of drugs and utilization of other services, which may be complements (a physician visit may be necessary to get a prescription) or substitutes (compliance with medications may reduce disease flare-ups and emergency visits). Evidence that even modest cost sharing for some chronic medications can significantly affect utilization of more costly medical services has generated great interest in ‘value-based insurance design,’ which would take these complementarities into account in designing cost sharing. Further research is needed into how optimal cost-sharing structures differ across disease states and drug types, and how their effects in practice are modified by stop-loss limits, manufacturer coupons, and other offsets.
Price And Reimbursement Regulation: The EU
In most industrialized countries with comprehensive insurance, payers control prices and utilization of biopharmaceuticals, with a view to maintaining access while managing within fixed health budgets. Price regulatory systems use three prototypical approaches to setting prices, and some countries use variants of multiple approaches.
Internal Referencing
Internal referencing compares the health outcomes with the new drug relative to one or more existing drugs and grants a price premium only if the new drug demonstrates superior safety, efficacy, or other benefits. In principle, this approach rewards innovation that produces measurable incremental value. It is usually applied only at launch. Postlaunch price increases are generally not allowed, and price decreases may be mandated if total expenditure for a drug exceeds the payer’s target based on the expected number of eligible patients. These ‘volume-price offsets’ reduce the price in proportion to the expenditure overrun. This not only keeps expenditure within target but also deters promotion beyond the target population.
A special case of internal referencing is ‘Bibliography: price reimbursement,’ as implemented in Germany and the Netherlands, in which the payer groups drugs based on similarity of indication, therapeutic effects, and sometimes mechanism of action. The Bibliography: price is the maximum reimbursement price for all drugs in the group, and if the actual price is higher, the patient must pay the excess. The Bibliography: price is usually based on a low-priced drug within the group, which could be a generic. If classes are broadly defined and ignore significant differences between drugs, this approach can undermine incentives for incremental innovation within a class. In Germany’s post-2010 approach to drug pricing, the first step is a formal review of the new drug, relative to comparators. If the new drug is deemed to offer no significant improvement it is assigned to a Bibliography: pricing group and is reimbursed at the prevailing Bibliography: price. If it is deemed significantly superior, then a new price is negotiated or determined by arbitration. Thus this approach recognizes the importance of benefit evaluation before assigning a drug to Bibliography: pricing.
External Referencing
With external referencing, the price of the new drug in country X is set at the mean, median, or minimum price of the same drug in a specified set of other countries. This approach is widely used in the EU, and the external Bibliography: may be the EU average price. This approach undermines the firm’s ability to maintain price differentials between countries although, as noted earlier, such differentials are consistent with Ramsey pricing principles applied to paying for the joint costs of R&D. Further, external referencing creates incentives for firms to delay or not launch drugs in small, low-priced countries, if these prices might undermine potentially higher prices in other countries. Several studies have found evidence of such delays and nonlaunch due to referencing within the EU. Thus, external referencing by one country can lead to spill-over reductions in access and presumably social welfare in referenced countries.
Parallel Trade
Although parallel trade is not a form of direct price regulation, it has effects similar to external referencing, but on a more limited scale. Parallel trade (also called commercial drug importation) permits commercial third parties – usually pharmacies and wholesalers – in one country to import drugs purchased in other, lower-priced countries, effectively arbitraging the price differences. The EU authorizes parallel trade between EU member countries as part of the general policy of free movement of goods within the EU.
Although economic theory generally concludes that free trade increases social welfare by enabling consumers to source products from lower cost producers and benefit from the savings, these conditions are generally not met for parallel trade in drugs. Price differentials for drugs between EU countries reflect differences in income and regulatory systems, not differences in production costs, hence there is no resource efficiency gain from such trade. On the contrary, parallel traded goods often require repackaging or relabeling which adds to resource costs. Further, the savings from arbitraging differences in exmanufacturer prices are largely captured by middlemen and are not transferred to consumers/payers. If the net effect of parallel trade is revenue redistribution from manufacturers to distributors that results in reduced incentives for R&D, then the efficiency effect of parallel trade is likely negative.
Cost-Effectiveness Review
An indirect approach to price control results when the payer reviews the incremental cost-effectiveness of a new drug, relative to standard of care, as a condition of reimbursement. The UK’s National Institute for Clinical Excellence exemplifies this approach, with detailed methodological requirements and an explicit threshold cost per QALY. Other countries, including Australia, Canada, and Sweden use similar approaches. If the manufacturer is permitted to set a price up to the maximum at which the new drug meets the ICER threshold, then this approach acts as an indirect control on price that rewards innovation and enables the manufacturer to capture the benefits produced, as required for dynamic efficiency, but without the payer having to directly regulate the price.
Conceptually, it is a simple step to convert cost-effectiveness analysis (CEA) review into an explicit value-based pricing (VBP) regime. VBP would allow a new drug a price premium over current treatment commensurate with its incremental value, which includes both incremental health benefits plus any cost savings. This VBP might be adjusted post-launch, if the evidence on incremental benefits changes. Whether the VBP should be adjusted if the price of the comparator changes due, for example, to generic entry, is an important policy question that requires further research.
Measurement Of Value
If payers are concerned to get maximum value from their expenditures on medical care, then measurement of value of health gain, using CEA and other approaches, is essential. CEA is used as part of broader health technology assessment (HTA) programs to evaluate the incremental health-related effects and costs of new technologies, including drugs, relative to existing technologies. This approach was adopted in the 1990s in Australia, New Zealand, the UK, and Canada, and variants have since been adopted in an increasing number of countries in Europe and more recently in Asia and Latin America. In the US, there is growing interest in comparative-effectiveness research, but with political reluctance to explicitly use cost per QALY or other outcome measures to make reimbursement decisions. CEA grew out of more general HTA, as payers sought more systematic, evidence-based approaches to resource allocation and adoption of costly new technologies within limited budgets.
Implementing value measurement raises both theoretical and practical issues that are being worked out as payers attempt to apply CEA to regulation of pharmaceutical use and prices. Practical questions include what types of evidence to use and how to deal with the inevitable gaps in evidence, especially at launch; use of riskor cost-sharing contracts when evidence is uncertain; and use of CEA as one among several criteria considered by decision makers. Considerable progress has been made over the past two decades in both theory and measurement of value, primarily using QALYs. Although many criticisms remain, similar and other criticisms are likely to apply to any alternative metric that attempts to provide a unidimensional measure of value that can compare outcomes across different health interventions. Until superior alternatives are developed, QALYs are likely to remain widely used.
Physician Dispensing
Pharmaceutical reimbursement raises unique issues in countries with physician dispensing. Japan, Taiwan, and South Korea have traditionally exemplified this approach, but each has recently taken steps to separate prescribing and dispensing, in contrast to China where most drugs are still prescribed and dispensed in hospitals and clinics. Simple economic theory and casual observation suggest that where physicians dispense the drugs that they prescribe and can profit from the margin between a drug’s acquisition cost and their reimbursement, manufacturers will offer discounts in order to increase this profit margin. The financial incentives of physicians may, therefore, lead to excessive prescribing and bias toward high-margin drugs. Japan traditionally mitigated this effect by biennial review of acquisition prices and downward revision of reimbursement prices to squeeze the margin.
Since 2000, Japan, South Korea, and Taiwan have all taken steps to encourage switching to pharmacy dispensing. The fundamental challenge is that if dispensing income is a significant fraction of total income for physicians, then payers are under pressure to increase other payments to physicians, in addition to now paying pharmacy dispensing fees, which may increase total expenditures. Japan took a gradual, incentive-based approach, paying increased prescription issuance fees for physicians and dispensing fees for pharmacists. The share of prescriptions dispensed through pharmacies has increased to more than 60% in 2011, but cost savings are uncertain because of the additional fees. Korea abruptly required that physicians cease dispensing drugs, which led to physician protests, increased fees, and apparently a shift to higher priced drugs. In response to physician protests, Taiwan allowed clinics affiliated with physician offices to continue dispensing as long as they hired a pharmacist and paid additional fees. Hence, again there has been no reduction in total medical expenditures. Thus, although the evidence suggests that physician prescribing does distort utilization, changing this is not easy and may lead to higher, not lower expenditures, at least in the short run.
Promotion
Biopharmaceuticals
Because the potential benefits and risks of pharmaceuticals are intrinsically nonobvious, providing information to physicians and consumers about a drug’s potential effects is critical to its appropriate use. Such information dissemination is provided and financed largely by pharmaceutical firms, through detailing of physicians, journal advertising, distribution of free samples, and direct-to-consumer advertising (permitted only in the US and New Zealand), subject to regulations that differ across countries. Estimates of the advertising-to-sales ratio in the US range from 6.7% to 18%. The highest estimates include samples valued at retail prices, which significantly overestimate the cost of samples to firms. High advertising-to-sales ratios reflect both the fact of multiple customers – physicians, patients, and payers – and the incentives created by inelastic demand resulting from extensive insurance coverage and high price-to-marginal cost ratios.
The economic literature on promotion is mainly from the US. It suggests that advertising may be both informative and persuasive, and both characteristics apply to some pharmaceutical advertising. Implications for public health and welfare depend on whether or how far advertising raises brand-specific versus industry-wide demand, impacts drug costs, and impacts competition and prices. Empirical evidence is mixed but suggests that consumer advertising is more effective at enlarging the general market, through more physician contact, expanded treatment, etc., whereas physician advertising is primarily persuasive, although the informative role is likely to be greater early in a drug’s lifecycle. There is no strong evidence that either consumer or physician-directed promotion raises prices. An overall welfare assessment would require a balancing of complex benefits and costs, and conclusions may depend on type of drug, stage of lifecycle, and other factors that affect the relative magnitude and value of information versus persuasion.
Medical Devices
Promotion of medical devices and equipment varies by sector, depending on the user/decision-maker, usually a hospital. However, for complex, implantable devices such as hips or stents, the surgeons who insert the devices may also be major customers because their ease of use with a device affects their time required and willingness to use a device. Such devices require promotion by technically qualified, skilled salespersons who may also play an important role in training the surgeons on how to use the devices. The empirical evidence suggests significant economies of scale in device marketing. This is plausible, because larger firms that produce a full range of products for a particular medical specialty, for example, orthopedics, can spread the fixed costs of hiring and training a dedicated salesforce that promotes only their products, whereas smaller firms that produce only one product may have to rely on general distributors who handle competitors’ products. Such economies of scale in marketing are plausibly one factor accounting for the general pattern that small-device firms with good products are usually acquired by larger firms, rather than attempting to seek external financing to grow as independent competitors. Comprehensive data on promotion, sales, and pricing are not available for devices as it is for drugs, hence this remains an important area for future research.
Emerging Markets: Self-Pay For Pharmaceuticals
Pharmaceutical markets in developing countries differ from those of industrialized countries in that insurance coverage for drugs is very limited, with most people paying directly out-of-pocket, especially those at lower income levels. Theory suggests that manufacturers might seek to practice price discrimination – charging lower prices in these countries than in higher-income countries – if they were assured that the drugs would not be exported to, or their lower prices would not be referenced by, higher-income countries. Similarly, price discrimination between rich and poor consumers within these countries would also increase sales for companies and access for consumers, if it were feasible. However, government policies, distribution systems, and other factors undermine market segmentation in developing countries, although corporate strategies such as dual branding, direct distribution to providers, and consumer coupons can be effective for some drugs. Inefficient distribution systems also pay a role in raising retail prices to consumers, regardless of prices charged by manufacturers in many developing countries.
The global nature of pharmaceutical R&D raises issues of appropriate cross-national price differentials to share the joint costs. Theoretical models of monopoly pricing using either price discrimination or uniform pricing and models of Ramsey pricing applied to payment for the joint costs of R&D suggest that differential pricing is welfare superior to uniform pricing across countries. Assuming that higher-income countries have more inelastic demand, this implies that richer countries should pay higher prices than poorer countries, and this is consistent with most norms of equity. The principle of differential pricing between the richest and poorest nations is widely accepted in policy debates. However, in practice, consensus breaks down on appropriate price differentials and absolute price levels, particularly for middle-income countries with emerging middle classes but large poor populations.
The evidence suggests that drug prices are higher, relative to average per capita income, in lowand middle-income countries. This applies to generics as well as on-patent drugs. Relatively high prices in low and middle-income countries partly reflects the highly skewed income distributions, which create incentives for firms to target the more affluent segment (Flynn et al., 2006). Further, because regulatory systems in these countries do not require that generic copies be bioequivalent to the originator, quality uncertainty leads producers to compete on brand, using both brand and high price as a proxy for quality (Danzon et al., 2011). In such markets, only the lowest-quality firms compete on price. However, regulatory requirements for bioequivalence of all generics would likely put many local firms out of business. Thus, the obstacles to reform are primarily political.
Vaccines
Preventive vaccines are biologics but differ from other biopharmaceuticals in important aspects. The external costs of infectious diseases imply external benefits from effective vaccines, and this has motivated public mandates, purchasing, and subsidies for vaccines in most countries and government subsidies to supply for particular products, such as Project Bioshield in the US. Relatively small market size and concentrated purchasing have contributed to the existence of few or sole suppliers of most individual vaccines in the US, which has resulted in shortages when the sole supplier experiences production problems.
A considerable literature has examined the cost-effectiveness of different vaccines in different contexts spanning both developed and developing countries, and appropriate policy responses to both suboptimal private demand and sole supplier markets. Policies to promote investment in vaccine R&D include push and pull incentives for the private sector, public production, and the no-fault Vaccine Injury Compensation Program that was implemented in the US in 1986.
After decades of being considered a neglected R&D sector, the past decade has seen a resurgence of interest in vaccines, with several large pharmaceutical companies and many smaller companies entering the US and EU markets, and several WHO-qualified suppliers of vaccines, from India and South Korea, now selling the majority of vaccines to emerging and middle-income countries. Thus, future research must consider factors that differentiate vaccines from other biologics and are common across all or most vaccines and market contexts versus factors that are specific to a particular vaccine or market context. The conditions for purchasing and supplying vaccines differ significantly across countries. Identifying these differences and their effects is a necessary part of generalizing about vaccine economics and appropriate vaccine policy.
Diagnostic Imaging
Like biopharmaceuticals, diagnostic imaging, including computed tomography, magnetic resonance imaging, positron emission tomography, and other technologies, poses challenges related to achieving appropriate use, pricing, and R&D incentives. However, the context and solutions are very different because these are durable machines with high fixed costs but low marginal cost to hospital or physician purchasers. Although a hospital may own the machine, the decision to order a scan is usually made by a physician who is not the same as the radiologist who interprets the scan and is reimbursed.
Conclusion
The biopharmaceutical and medical equipment industries pose many interesting economic questions that are different from the textbook economic industries or the health services sectors. Like health services, the role of insurance is fundamental in affecting demand. However, because these are research-intensive industries, optimal insurance and reimbursement design must consider effects on producers’ incentives, short and long run, as well as effects on consumer protection. Much progress has been made in understanding the economics of R&D, effects of regulation, promotion, and pricing and reimbursement, particularly for biopharmaceuticals. But this remains a fertile field for future research.
Bibliography:
- Danzon, P. M. and Furukawa, M. (2011). Cross-national evidence on generic pharmaceuticals: Pharmacy vs. physician-driven markets. NBER Working Paper 17226. Cambridge, MA: NBER.
- Danzon, P. M., Mulcahy, A. and Towse, A. (2011b). Pharmaceutical prices in emerging markets: effects of income, competition and procurement. NBER Working Paper 17174. Cambridge, MA: NBER.
- Danzon, P. M., Towse, A. and Mestre-Ferrandiz, J. M. (2011a). Value-based differential pricing: Efficient prices for drugs in a global context. NBER Working Paper w18593. Cambridge, MA: NBER.
- Di Masi, J. and Grabowski, H. (2007). The cost of biopharmaceutical R&D: Is biotech different? Managerial and Decision Economics 28(4–5), 285–291.
- Ernst and Young. (2012) Pulse of the industry: Medical technology report. New York: Ernst and Young.
- Flynn, S., Hollis, A. and Palmedo, M. (2009). An economic justification for open access to essential medicine patents in developing countries. Journal of Law Medicine and Ethics 37(2), 184–208.
- Garber, A., Jones, C. I. and Romer, P. M. (2006). Insurance and incentives for medical innovation. Forum for health economics and policy: Vol. 9, Issue 2 Article 4. Cambridge, MA: Biomedical Research and the Economy.
- Mestre-Ferrandiz, J. M., Sussex, J. and Towse, A. (2012). The R& D cost of a new medicine. London: Office of Health Economics.
- Claxton, K., Briggs, A., Buxton, M. J., et al. (2008). Value based pricing for NHS drugs: An opportunity not to be missed? British Medical Journal 336, 251–254.
- IMS Market Prognosis (2012). Report of the IMS Institute of Healthcare Informatics. Available at: www.ims.com (accessed 20.03.13).
- Lakdawalla, D. and Sood, N. (2009). Innovation and the welfare effects of public drug insurance. Journal of Public Economics 93, 541–548.
- Malueg, D. and Schwartz, M. (1994). Parallel imports, demand dispersion, and international price discrimination. Journal of International Economics 37, 167–195.