Although the biotech industry is a relatively new source of medical therapies – its first new drug approvals came in the early 1980s – it has recently become a major source of drug industry growth and innovation. New biological entities (NBEs) have a significantly higher likelihood of being a first-in-class or novel introduction compared with other new drug entities (Grabowski and Wang, 2006). For example, the oncology class has experienced the introduction of breakthrough monoclonal antibodies and targeted biological agents resulting from increased knowledge of the molecular mechanisms for cancer (DiMasi and Grabowski, 2007a). Substantial improvements in survival, morbidity, and patients’ quality of life have been documented in diseases previously resistant to successful treatment, such as aggressive HER-2 positive breast cancer (Smith et al., 2007) and disability associated with rheumatoid arthritis (Weaver, 2004).
Although NBEs have been an important source of biopharmaceutical innovation, they have also accounted for a rising share of overall drug expenditures in the US and worldwide. They now account for approximately one-quarter of all the US expenditures on pharmaceuticals and represent approximately half of all products in clinical testing (Trusheim et al., 2010). NBEs for oncology patients and other indications also can cost tens of thousands of dollars per course of treatment. They are also frequently targeted to life-threatening and disabling diseases. These facts and trends have made biological entities an increasing focus of attention for policymakers and payers grappling with rising healthcare costs and budgets.
A recent development in Europe and the US is the establishment of an abbreviated pathway for the so-called biosimilars – biological products that are similar to, but not identical with, a reference: biological product in terms of quality, safety, and efficacy. Biologics are typically more complex molecules than small-molecule chemical drugs. Biologics are manufactured not through chemical synthesis but through biological processes involving manipulation of genetic material and large-scale cultures of living cells, where even small changes to the manufacturing process may lead to clinically significant and unintended changes in safety and efficacy. As a result, establishing that a biosimilar is ‘similar enough’ to achieve comparable therapeutic effects in patients is a much more challenging task for companies and regulators than establishing bioequivalence for generic chemical drugs. Biosimilars generally require analytical studies, animal testing data, and some clinical trial evidence on safety and efficacy to gain approval. Biosimilars can provide an important new source of competition to established biological entities. A key issue at the present time is how this competition is likely to develop and how it will influence expenditures for biopharmaceuticals by payers and consumers, investment in innovation, and the research, development, and marketing processes for manufacturers.
The EU has had a framework in place for approving biosimilars since 2005. The European Medicines Agency (EMA) has issued general and class-specific guidelines in six classes and has approved biosimilars in three product classes – somatropins, erythropoietins, and granulocyte colony-stimulating factors (G-CSFs). The experience of biosimilars in various European countries is considered later in this article.
In March 2010, as part of the overall Patient Protection and Affordable Care Act, the US Congress created an abbreviated pathway to approve biosimilars. The Food and Drug Administration (FDA) is in the process of implementing the law, including consulting with potential entrants and developing and releasing for public comment draft guidelines. The US situation is of particular interest as it has been the center of biotech innovation and the country with the largest expenditures on biological products. Although the US has a strong history of generic drug utilization, until the 2010 Act, there was no corresponding pathway for biosimilar entry.
In this article, the authors first discuss regulatory, reimbursement, and economic factors that will affect how competition between branded biologics and biosimilars may evolve. These factors are based on current market dynamics including initial European biosimilar experiences, the provisions of the new US law enacted in 2010, and the US experiences under the Hatch-Waxman Act. Taking into account the scientific, manufacturing, and other differences between biologics and chemically synthesized drugs, and between the regulatory frameworks governing each, expected biosimilar competition is then compared and contrasted with generic competition. Finally, the likely impact of biosimilars on cost savings is briefly assessed and potential impacts on innovation incentives in the biopharmaceutical industry is discussed.
Biosimilar Experience In The European Union
The EU has had in place a well-defined regulatory pathway for biosimilars for several years. In October 2005, the European Commission adopted an EMA framework for the approval of biosimilars. The framework includes an overarching set of principles; general guidelines on quality, safety, and efficacy; and guidelines specific to product classes. To date, the EMA has issued guidelines in six therapeutic classes. Guidance is under development for three other major types of biologics: monoclonal antibodies, recombinant follicle-stimulating hormone, and recombinant interferon beta. Other countries have used a European-like approach, including Canada (where biosimilars are termed ‘subsequent entry biologics’ (SEBs)) and Japan. Australia adopted the EU guidelines in August 2008.
The EMA has required at least one Phase II or III clinical trial for biosimilars to demonstrate similar safety and efficacy to their References: molecules. As opposed to the legislative biosimilar framework in the US, in which the FDA approves applications as biosimilars or interchangeable biosimilars, the EMA framework does not result in any findings of interchangeability, and questions of substitution are left to the member states to regulate. Local substitution laws differ across the EU member states, with some including explicit prohibitions on automatic substitution for biologics (such as Spain and France).
Since 2006, 14 biosimilar products in three therapeutic classes – erythropoietins, somatropin, and granulocyte colony-stimulating factors (G-CSFs) – have been approved, referencing four innovative products, and 13 are currently marketed in Europe. Three applications for biosimilar human insulin (with different formulations) were withdrawn in December 2007, based on failure to demonstrate comparability, and one approved product was later withdrawn (Table 1).
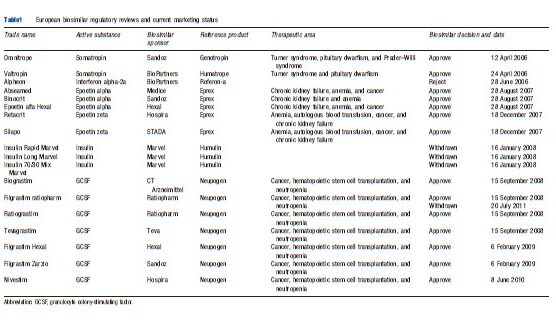
Empirical Evidence From Biosimilars In The European Union
Germany has exhibited the highest level of aggregate demand and market share for any biosimilar product (erythropoietin). To date, Germany’s Federal Healthcare Committee, which decides which products and services are reimbursed, has embraced biosimilars wholeheartedly. In addition to a References: pricing system in place for biosimilars, Germany has specific targets or quotas for physician and sickness funds for biosimilars that vary by region. Furthermore, Germany is a main source of biosimilar manufacturing in Europe, and biosimilar companies generally enjoy strong reputations with healthcare providers.
Uptake in other European countries has been slower. In some cases, this reflects later biosimilar entry dates and the timing of reimbursement approval by government payers. Although evidence from experiences in Germany or other European countries with biosimilar substitution are not directly applicable to other markets, given differences in the markets and pricing, access, and reimbursement systems, they nevertheless suggest that over time, payers, physicians, and patients will accept biosimilars.
Table 2 summarizes biosimilar shares in five large European countries: France, Germany, Italy, Spain, and the UK, for the therapies somatropin, erythropoietin alpha, and G-CSF from 2007 to 2009. The extent of biosimilar penetration varied substantially both across therapies within a country and across countries for the same therapy. In Germany, the biosimilar erythropoietin alpha accounted for 62% of total biosimilar and innovator product units sold in 2009, within 2 years of its launch; by contrast, in France, Italy, Spain, and the UK, biosimilar erythropoietin alpha had less than a 5% share in 2009. Biosimilar market shares for G-CSF in 2009 ranged from 21% (UK) to 7% (Spain). However, there is evidence that biosimilar G-CSF shares have grown rapidly in several European countries since 2009 (Grabowski et al., 2012). In particular, a study undertaken by IMS Health found that biosimilars in the G-CSF class had shares more than 50% in Germany, France, and the UK by the third year after launch, and characterized the market for this class in these counties as being commodity-like and mainly controlled by payers (IMS, 2011a). In contrast, the shares for somatropin are lower than the other two classes in most European countries, reflecting conservative physician prescribing and a differentiated market with competition based on price, promotion, and delivery device-based patient convenience.
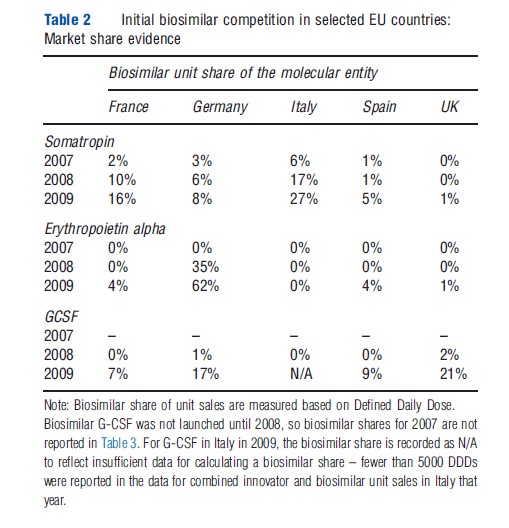
Biosimilar market development (and share uptake) may differ between European countries and the US, given the differences between their healthcare systems. For example, the US is more litigious than Europe; thus, the FDA may decide to proceed more cautiously and require more clinical data than the EMA has in the past. This broad generalization may not always hold true; however, in the US, the FDA approved Sandoz’s enoxaparin sodium abbreviated new drug application (ANDA) as a fully substitutable generic (referencing Lovenoxs) requiring no clinical evidence. In contrast, the EMA requires clinical data to approve a biosimilar application for a low molecular weight heparin. Future research comparing biosimilar market attitudes and experience in European countries, countries with a European-like approach (e.g., Australia, Japan, and Canada), the US, and other nations (e.g., the so-called ‘BRIC’ nations of Brazil, Russia, India, and China) is needed. Given the significant differences in the regulatory, medical delivery, and reimbursement systems between less-developed and more-developed nations, the pattern of biosimilar competition may also be very different.
The United States Biologics Price Competition And Innovation Act
The Biologics Price Competition and Innovation Act of 2009 (BPCIA), enacted as part of the Patient Protection and Affordable Care Act of 2010 (PPACA), created an abbreviated pathway for the FDA to approve biosimilars. This legislation complements the 28-year-old Drug Price Competition and Patent Term Restoration Act of 1984 (generally referred to as the Hatch-Waxman Act), which provides a clear path for generic drug entry in the case of new chemical entities (NCEs) approved under the Food, Drug, and Cosmetic Act (FD&C Act) through the ANDA process. Through that process, generic drugs demonstrated to be bioequivalent to off-patent References: drugs may be approved without the submission of clinical trial data on efficacy and safety. ANDA approval requires a finding that the generic drug is bioequivalent to its References: drug and has the same active ingredient(s), route of administration, dosage form and strength, previously approved conditions of use, and labeling (with some exceptions). Some initially marketed biologic products were approved under the FD&C Act, such as human growth hormones. However, most large molecule biologic medicines were approved under the Public Health Service Act and have not been subject to generic competition under the ANDA process of the Hatch-Waxman Act. Biologic medicines approved under the Public Health Service Act will now be subject to competition from products coming to market through an expedited biosimilar approval process – relying at least in part on the innovator’s package of data or a prior FDA approval – for the first time as a result of the BPCIA.
The key provisions of the new legislation establishing an abbreviated pathway for the FDA to approve a biosimilar are:
- Biosimilarity: A biosimilar does not have to be chemically identical to its References: product but must be ‘‘highly similar to the References: product notwithstanding minor differences in clinically inactive components’’ and there must be ‘‘no clinically meaningful differences, in terms of safety, purity, and potency.’’ (PPACA, Section 7002 (b)(3))
- Interchangeability: The FDA may deem a biosimilar interchangeable with its References: product if it can be shown that it ‘‘can be expected to produce the same clinical result as the References: product in any given patient’’ and that ‘‘the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the References: product is not greater than the risk of using the References: product without such alternation or switch.’’ (PPACA, Section 7002 (k)(4)) The first biosimilar shown to be interchangeable is entitled to a 1-year exclusivity period during which no other product may be deemed interchangeable with the same References: product.
- Regulatory review: The FDA will determine whether a product is biosimilar to a References: product based on stepwise consideration of analytical, animal-based, and clinical studies (including the assessment of immunogenicity and pharmacokinetics or pharmacodynamics). In February 2012, the FDA released the first three documents in a set of guidance documents for the development of biosimilars under BPCIA.
- Regulatory Exclusivity for the innovative biologic: Biosimilar applications may be submitted beginning 4 years after FDA approval of the References: innovative product. Before the FDA can approve a biosimilar using the abbreviated pathway, there is a 12-year period of exclusivity following FDA approval of the innovative biologic. An additional 6 months of exclusivity is available for the References: innovative biologic if pediatric-study requirements are met, which applies to both the 4and 12-year exclusivity periods. The most important (and contentious) of these exclusivity provisions is the 12 years of exclusivity for an innovative biologic before a biosimilar can enter using an abbreviated application. This 12year exclusivity term is referred to as regulatory exclusivity in distinction from the exclusivity afforded through patents granted by the US Patent and Trademark Office.
- Limitations on 12-year exclusivity: Several types of licensures or approvals are not eligible for 12-year exclusivity, including: (1) a supplemental biologics license application (sBLA) for the References: biologic; (2) a subsequent BLA filed by the same sponsor, manufacturer, or other related entity as the References: biologic product that does not include structural changes in a biologic’s formulation (e.g., a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device, or strength); or (3) a subsequent BLA filed by the same sponsor, manufacturer, or other related entity as the References: biologic product and that includes structural changes in a biologic’s formulation but does not result in improved safety, purity, or potency.
- Reimbursement: A potential disincentive for biosimilar adoption is mitigated by setting the reimbursement for a biosimilar under Medicare Part B at the sum of its Average Selling Price (ASP) and 6% of the ASP of the References: biologic.
- Patent provisions: The BPCIA requires a series of potentially complex private information exchanges between the biosimilar applicant and References: product sponsor, followed by negotiations and litigation, if necessary. In contrast to the patent provisions for NCEs under the Hatch-Waxman Act, there is no public patent listing akin to the Orange Book, no 30-month stay when a patent infringement suit is brought, and no 180-day exclusivity awarded to the first firm to file an abbreviated application and achieve a successful Paragraph IV patent challenge.
Food And Drug Administration Regulations And The Costs Of Developing A Biosimilar
The new law authorizing biosimilars gives broad latitude to the FDA to define the process and standards for approval. FDA decisions will affect both the demand for and the supply of biosimilars:
- The level of evidence required will affect the costs of market entry, the number of biosimilar entrants, and the assets and capabilities required to compete successfully.
- The level of clinical trials and other evidence required to establish interchangeability or similarity will also potentially affect the level of market adoption, as greater levels of evidence may increase physicians’, payers’, and patients’ confidence in a biosimilar medicine.
- Naming conventions and pharmacovigilance requirements for biosimilars will affect market entry and perceptions of substitutability by physicians, payers, and patients, as well as safety monitoring after launch.
- Whether data on one indication can be extrapolated to others – absent additional clinical trials in that patient population will have an impact on entry decisions, perceptions of substitutability, and biosimilar market uptake.
- Definitions of what constitutes changes in ‘safety, purity, or potency,’ as they are applied to determine whether a 12-year exclusivity is to be authorized for next-generation products will affect biotech investor incentives.
Criteria For Establishing Biosimilarity
The initial draft guidance documents released by the FDA in February 2012 state that ‘‘FDA intends to consider the totality of the evidence provided by a sponsor to support a demonstration of biosimilarity’’ (emphasis added). For a given biosimilar application, the FDA draft guidance notes that ‘‘(t)he scope and magnitude of clinical studies will depend on the extent of residual uncertainty about the biosimilarity of the two products after conducting structural and functional characterizations and possible animal studies.’’ (Food and Drug Administration (FDA), 2012a, pp. 2, 12). Theoretically, this could encompass, at one extreme, only a bioequivalence study (similar to what is required for generic approval under Hatch-Waxman) or, at the other extreme, when science and experience require more data, a full program of clinical studies equivalent to that included in a biologic’s license application.
FDA officials, in a New England Journal of Medicine publication, had previously stated that ‘‘[a]lthough additional animal and clinical studies will generally be needed for protein biosimilars for the foreseeable future, the scope and extent of such studies may be reduced further if more extensive fingerprint-like characterization is used.’’ (Kozlowski et al., 2011, p. 386) In the future, the agency hypothesizes, the current state-of-the-art for analytic characterizations may advance to allow highly sensitive evaluations of relevant product attributes and permit a ‘fingerprint-like’ identification of very similar patterns in two different products (such strategies were cited in the FDA’s approval of the Sandoz ANDA for enoxaparin sodium, a complex mixture, mentioned later in this article.)
The costs of an FDA submission for the US approval could be lower for biosimilars already on the market in Europe if the biosimilar can rely on previously undertaken European clinical trials, at least for some products. In its draft guidance documents released in February 2012, the FDA noted it will accept clinical studies undertaken for approval in other jurisdictions under certain circumstances, when justified scientifically and when accompanied by ‘bridging’ data. However, it also noted, ‘‘[a]t this time, as a scientific matter, it is unlikely that clinical comparisons with a non-US-licensed product would be an adequate basis to support the additional criteria required for a determination of interchangeability with the US-licensed References: product,’’ and the specific data requirements for products will be determined by the FDA on a case-by-case basis. (Food and Drug Administration (FDA), 2012b, p. 8.)
If the FDA requires significant clinical trial evidence, approvals for biosimilars, as compared with generics, will require a much bigger investment. The cost for biosimilar approval will depend on the number and size of the necessary clinical trials, the number of indications involved, and other specific FDA requirements. The current requirement for a BLA is typically two large-scale Phase III pivotal trials. If the FDA requires at least one Phase II/III type study comparable to those undertaken by innovators, then the out-of-pocket costs will likely be in the range of US$20 million to US$40 million for the studies alone. In addition, the preclinical costs associated with biosimilars may in some cases be higher for biosimilars than for innovative products, as they entail modifying the production process to achieve a specific profile that very closely approximates the References: product without the benefit of the innovator’s experience. Others have estimated that for very complex biologics such as some monoclonal antibodies, biosimilar development costs could total US$100 million to US$200 million and take 8 or more years to bring a product to market (Kambhammettu, 2008). In contrast, the cost of completing bioequivalence studies for generic drugs is estimated to be only US$1 million to US$2 million.
Regulatory Requirements For An Interchangeability Designation
Another key regulatory issue will be the analytical and clinical evidence required to deem a biosimilar interchangeable with its References: product, thus enabling automatic substitution without physician approval, subject to relevant state laws. Under the BPCIA, for products used more than once by patients (the majority of biologics), the biosimilar sponsor will need to demonstrate that switching between the biosimilar and References: product poses no additional risk of reduced safety or efficacy beyond that posed by the References: product alone. Postapproval interchangeability assessments may require a strong postmarketing system and evaluation of postmarketing data.
Achieving an FDA finding of interchangeability may be associated with far greater development costs than achieving a determination of biosimilarity, so it may be limited initially to a select few examples where molecules meet certain tests for establishing ‘sameness’ through differentiated characterization or other available technology. For instance, the availability of differentiated analytical characterization technology supported the FDA’s approval of Sandoz’s ANDA for generic enoxaparin sodium (referencing Lovenoxs). Although not a biosimilar (Lovenoxs, a chemically synthesized product derived from natural sources, has been described as a complex mixture), the factors that the FDA cited in its approval may give some insight into the Agency’s current approach and how continued technological change could influence the evidence necessary to establish interchangeability in the future.
For classes of more complex biologics, applications for biosimilarity will likely require some clinical trial data in order to be approved and costly switching trial data in order to be deemed interchangeable. Many firms may elect not to make the investments necessary to pursue interchangeability initially, given the current state of scientific knowledge regarding biosimilars and high levels of regulatory uncertainty. This is in contrast to small-molecule generic drugs, where an ‘A’ rating by the FDA recognizes the products as therapeutically equivalent and eligible for substitution by pharmacists without physician approval, subject to state substitution laws, thus driving rapid share loss by the branded References: product.
Manufacturing Costs
The ongoing cost of manufacturing biological entities is also significantly higher than for chemical entities. Biosimilar manufacturers may need to construct expensive plants or obtain long-term lease or purchase agreements with third parties that have an FDA-approved facility if they do not already have excess suitable manufacturing capacity. In any event, the cost of entry for biosimilars in terms of plant capacity is likely to be an order of magnitude higher than for generic drug products (which may total only US$1 to US$2 million) and may be closer to two orders of magnitude higher. The high costs of entry – particularly the substantial capital requirements – are likely to restrict the number and types of biosimilar entrants, at least initially. Furthermore, initial entry is likely to be limited to the biologics with the largest revenues and those where scientific and market feasibility have been demonstrated in Europe.
The Perspectives Of Healthcare Payers, Providers, And Patients
Reimbursement And Payer Considerations
Payer reimbursement policies and access control mechanisms also can substantially affect the extent and speed of biosimilar uptake. Consistent with relevant local laws, regulations, and practices, payers will develop coverage and reimbursement policies and make individual pricing, reimbursement, and access decisions for biosimilars and their branded References: products.
Cost sensitivity and willingness to encourage the use of biosimilars in place of their References: therapies may vary across different payers, including private insurers and public payers. Payer controls that restrict patient and physician therapy choice and access may also vary according to the setting in which care occurs (e.g., inpatient hospital or physician office), whether the biosimilar is rated interchangeable, the therapeutic indication and disease severity (e.g., oncology or growth disorders), as well as other factors.
Private insurers
Historically, in the US, managed care plans have been reluctant to restrict access or pursue aggressive cost-control measures because many biologic therapies are: (1) targeted to life-threatening illnesses such as cancer or other diseases that involve serious disability and (2) often lack close substitutes. In addition, biologics that are dispensed by physicians are often managed within plans as medical benefits rather than pharmacy benefits and are typically less subject to centralized controls or formulary restrictions. This has been changing over the past several years, particularly in indications where there is a choice between multiple brand name biologics. The introduction of biosimilars can be expected to accelerate these trends toward more active management of biologic choice, costs, and utilization.
Medicare
Medicare reimburses biologics under either the Part B or the Part D program, depending on the mode of administration. Many biologic drugs are currently dispensed in a physician’s office, clinic, or hospital as infused agents. The use of these biologics for Medicare patients is covered under the Medicare Part B program, whereas self-injectable biologics dispensed in pharmacies (including by specialty pharmacy or mail-order programs) are covered by the Part D program.
Medicare Part B
In designing the new abbreviated pathway for biosimilars, Congress acknowledged that the Medicare rules for reimbursement of drugs administered under Part B could provide inadequate financial incentives for providers to utilize lower priced biosimilars. Part B drugs have historically been purchased through a ‘buy and bill’ approach by providers who also make decisions about which therapies are appropriate for a given patient. The provider is reimbursed by Medicare for administering a Part B drug, and the level of reimbursement is based on the manufacturer’s weighted ASP for the category to which the drug belongs (defined by a unique code), plus 6%. When generics are assigned to the same code as their References: new chemical entity, physicians receive the same level of reimbursement, the volume-weighted average ASP for all manufacturers’ products, for using either the generic or the References: product. Thus, physicians generally have a strong incentive to utilize the lower cost generic product, (although the physician’s choice of generic or References: product also depends on the net acquisition cost of both products to the physician, based on any contracts that may be in place with the brand manufacturer as well as the pricing strategy of the generic entrant).
Because biosimilars are unlikely to be deemed interchangeable by the FDA, at least initially, to the degree they are thus unlikely to be assigned to the same code as the brand product, physicians may have an incentive to utilize the more expensive (higher ASP) References: product for patients, as reimbursement is based on ASP plus 6%. To mitigate potential financial disincentives for physicians to adopt biosimilars, the new legislation sets biosimilar reimbursement under Medicare Part B at the sum of the biosimilar’s ASP and 6% of the ASP of the References: biologic product. The References: biologic product will continue to be reimbursed at its own ASP plus 6%. By basing the 6% payment to providers on the References: brand’s ASP, the legislation seeks to mitigate provider disincentives to adopt lower cost biosimilars when they are not deemed to be interchangeable and are placed in separate codes. Whether this reimbursement provision will be sufficient to overcome physician experience and loyalty to the References: biologic, as well as other financial incentives, is an open question.
Medicare Part D
Privately offered Medicare Part D drug programs cover drugs available at retail or via mail order, including selfinjectable biologics. Biologics accounted for only 6% of total prescription drug costs in the Medicare Part D program in 2007 (Sokolovsky and Miller, 2009); however, spending for biologics within the Part D program is expected to increase rapidly in the coming years. Between 2007 and 2008, MedPac estimates indicate that prices paid for drugs on specialty tiers (including biologics) in the Part D program grew by 18%, compared with 9% for all Part D drugs. Expenditures for selfinjected biologics are expected to continue to grow rapidly as these agents are increasingly used to treat a range of diseases, from rheumatoid arthritis to multiple sclerosis to human growth deficiency, and a large number of new biologics are currently under development. The high price of self-injected biologics relative to traditional NCEs also suggests that biologics will comprise an increasing share of Part D expenditures. This shift may lead payers to pursue pharmacy management techniques aimed at controlling utilization of these biologics.
Many Medicare Part D plan designs include a specialty drug tier, with median coinsurance rates increasing from 25% in 2006 to 30% in 2010 for stand-alone prescription drug plans and to 33% in 2010 for drug plans offered as a part of Medicare Advantage (Hargrave et al., 2010). Coinsurance plan designs could produce strong incentives to utilize biosimilars if substantial discounts emerge for biologic products with expensive courses of treatment for patients. Preferred specialty drugs might be subject to lower rates of coinsurance, to a copayment rather than to coinsurance, or to lower patient out-of-pocket costs at the same coinsurance rate.
One limiting factor to formulary incentives for biologics in Medicare Part D is that enrollees with low-income subsidies make up a disproportionately large share of the market for biologics under the Part D program. Given that these individuals are subject to limited cost sharing, other instruments such as step therapy and prior authorization may be employed to provide incentives for the use of biosimilars.
Medicaid
Medicaid Preferred Drug Lists (PDLs) reflect preferred biologic products in a number of therapeutic categories. Preferred drugs can be dispensed without the access controls (e.g., prior authorization) applied to nonpreferred drugs. For example, online PDLs for Florida, Illinois, New York, Ohio, Pennsylvania, and Texas indicate that rheumatoid arthritis (RA), hepatitis C (HCV), and human growth hormone formularies in these six large states preferred two or three RA agents (of six), one or two HCV agents (of five), and between two and five human growth hormones (of nine agents/forms). Medicaid programs can be expected to encourage biosimilars through PDLs and other medical management instruments. States with managed Medicaid programs apply formulary and access management techniques common in commercial insurance plans, and such managed programs are becoming more common.
Hospitals
Hospitals typically bear the costs of all drugs, including biologics, used during inpatient hospital stays as part of a fixed diagnosis-related group-based reimbursement per admission (DRG) that includes all services and products used during the episode of care. Consequently, these hospitals have incentives to implement formularies of preferred drugs and other mechanisms that encourage the use of lower priced products, possibly including biosimilars. As a result, for biologics that are generally used in hospital settings, hospitals will play a larger role than insurance companies in determining the demand for biosimilars. In the hospital sector, Pharmacy and Therapeutics (P&T) committees review the drugs that are stocked, on standing order forms, and which can be used by physicians. Hospitals also rely on Group Purchasing Organizations (GPOs) to gain leverage in negotiating discounts from suppliers, including biologic manufacturers. Because the hospital GPO market is highly concentrated, favorable contracts with a handful of suppliers can affect product selection. In addition, fixed reimbursement creates strong incentives for input cost reductions. To the degree that biologics used in the inpatient hospital setting are included in the DRG, depending on how significant a portion of spending they represent, hospitals may be more aggressive in implementing access controls to favor the utilization of some biosimilars, if biosimilar prices are not countered by originator manufacturer discounts.
United States Healthcare Reform Initiatives
More widespread adoption of comparativeand cost-effectiveness analyses across the US healthcare system could also influence adoption of biosimilars. Formal cost-effectiveness reviews by payers have been well established in countries outside the US in the form of Health Technology Assessments (HTAs). In the UK, for example, the National Institute for Health and Clinical Excellence’s (NICE) coverage recommendations have been based on strict reviews of cost-effectiveness calculations relative to current treatment, with an implied threshold value of an acceptable incremental cost per quality-adjusted life-year (QALY).
Finally, long-term changes in reimbursement policies may also shift financial incentives toward the use of biosimilars. For example, the adoption of global payment strategies, rather than fee-for-service reimbursement, or some form of shared savings, could strengthen the link between physician and/or hospital compensation and the use of lower priced biologics. Global payment strategies provide incentives for the adoption of lower cost treatments (and potentially encourage greater price competition) by setting a fixed payment level for a patient/episode of care, with all or some portion of the cost savings accruing to the care providers. Several states are considering implementing global payment strategies, and it has been suggested that government programs such as Medicaid could be the first to implement these strategies.
Patient And Physician Perspectives
The rate of biosimilar penetration is expected to vary by disease indication, patient type, physician specialty, and other factors. As noted, rates of patient and physician acceptance of biosimilars are expected to be lower when the biosimilar lacks an interchangeability rating. In addition, rates of biosimilar acceptance may vary according to such physician and patient focused factors as: Whether the physician specialty is historically more price-sensitive or demonstrates greater levels of brand loyalty in therapy choice (for instance, allergists vs. rheumatologists); whether the biosimilars will be used long-term as maintenance therapy or only once or twice (particularly if long-term clinical data are not available); whether the indication is life threatening or the implications of therapeutic nonresponse or adverse reactions are perceived to be very serious; or whether the difference in ease-of-use or out-of-pocket cost to the patient of the brand instead of the biosimilar is expected to be high.
When patients are stable on a given maintenance therapy, biosimilar substitution may tend to be concentrated among new patient starts. (The same is true of ‘switches’ between one branded drug and another.) As a result, the penetration of biosimilars for indications with a low rate of turnover in the patient populations may be limited if products are not interchangeable. The degree of biosimilar uptake will also depend on cost differences and the financial incentives to utilize biosimilars employed by managed care and government payers. These incentives, however, are likely to be tempered if existing patients are responding well to an established therapy. Other factors such as specialists’ brand loyalty, clinically vulnerable patient populations, and physician conservatism in switching stable patients to new therapies are also likely to keep rates of biosimilar uptake for current patients below those for new patients.
Another important demand-side factor is the perspective of specialist physicians and patient groups concerning biosimilars. Physicians who have years of experience with the References: biologic may be reluctant to substitute a biosimilar even for new patients until sufficient experience has been accumulated in clinical practice settings, as opposed to in clinical trials. To stimulate demand, it may be necessary for biosimilar firms to establish ‘reputation bonds’ with physicians through strategies similar to those employed by branded firms that communicate information to establish brand value through physician detailing, publications, advertising, and education programs. In addition, patient assistance programs and contracts with health plans, pharmacy benefit managers (PBMs), hospitals, or provider groups, which exercise control over therapy choice, may be used in a targeted way to affect the economic proposition associated with biosimilar adoption. These measures will increase the cost of drug distribution and marketing for biosimilars compared with small-molecule generic drugs, where such marketing and sales costs are minimal and demand is purely driven by lower price and pharmacy contracts for availability.
Biosimilar Competition Versus Generic Competition
Since the passage of Hatch-Waxman 28 years ago, generic entry has become a principal instrument of competition in the US pharmaceutical market. Generic products in 2010 accounted for 78% of all the US retail prescriptions, (IMS, 2011b) compared with only 19% in 1984 (Federal Trade Commission, 2002). As discussed, the growth of generic utilization has been accelerated by various formulary and utilization management techniques such as tiered formularies, prior authorization and step therapy requirements, higher reimbursements to pharmacies for dispensing generics, and maximum allowable cost (MAC) programs.
A distinctive pattern of generic competition has been observed in numerous economic studies (Grabowski, 2007). There is a strong positive relationship both between a product’s market sales and the likelihood of a patent challenge and between the number of generic entrants and the intensity of generic price competition once the exclusivity period has expired. An increasing number of products are now subject to patent challenges earlier in their product life cycle, as generic firms seek out the 180-day exclusivity period awarded to the first firm to file an ANDA with a successful Paragraph IV challenge. Successful products typically experience multiple entrants within the first several months after patent expiration, and generic price levels drop toward marginal costs rapidly as generic entry increases.
Theoretical Models Of Biosimilar Competition
Given the much higher costs of entry for biosimilars compared with generic drugs, as well as the other demandand supplyside factors discussed in the section Food and Drug Administration Regulations and the Costs of Developing a Biosimilar, the pattern of biosimilar competition is expected to differ from current generic competition. In particular, fewer entrants and less intensive price discounting are expected and competition may resemble branded competition more than generic competition (Grabowski et al., 2006). This is currently the case in the human growth hormone market, where eight products compete both through price, patient support, and product delivery differentiation. In 2006, Sandoz entered the human growth hormone market with Omnitropes (which References:d Pfizer’s Genotropins, via the section 505(b)(2) pathway of the Hatch-Waxman Act). Omnitropes has struggled to gain market share. Initially, it was reported to have priced at a 30% discount based on wholesale acquisition cost (WAC) compared with the most widely used biologic in this class, Genotropins. By 2008, Omnitropes’s discount had increased to 40% (Heldman, 2008). Despite these discounts, its share of somatropin use remained below 5%. These outcomes may not be reflective of the pattern of substitution for biosimilars generally, given that the human growth hormone market was a mature one with a number of competitors, and also given the differentiation by established brands via sophisticated penor needle-free delivery systems in this product class. With the approval of a pen delivery device system, and a strategy that includes physician detailing and patient support services, Omnitropes’s share of prescriptions dispensed increased to 19% in September 2012.
To date, some theoretical analyses have attempted to model the likely scenarios for biosimilar competition in the US market. One paper implements a simulation approach and projects that the relatively high cost of biosimilar entry will result in relatively small number of entrants even for larger selling biologic products and more modest discounts on biosimilars than in the case of generics (Grabowski et al., 2007). Other research relies on a segmented model of biosimilar competition, where biosimilars would be utilized significantly in price-sensitive segments of the market but less so in the nonprice-sensitive segments (given the reluctance of many providers to utilize biosimilars until considerable clinical experience has accumulated) (Chauhan et al., 2008). In this model, average price discounts depend on the relative size of these market segments. The findings indicate that, given a relatively small number of branded biosimilar competitors, the innovator will discount prices from preentry levels but not as much as the biosimilar entrants. This is in contrast to generic competition where branded firms typically do not lower prices postentry but may license an authorized generic when only a small number of generic competitors are expected as a result of a successful paragraph IV entry with a 180-day exclusivity award (Berndt et al., 2007).
Empirical Studies Of Generic Drug Analogs
Another line of research attempts to predict how biosimilar competition will emerge by considering analogous situations, including the US generic market for certain products which share some characteristics suggestive of biologics. In one example of this research, small-molecule drugs are divided into two classes, noncomplex and complex, with complex drugs being those that meet two of the following criteria: black box warnings, narrow therapeutic index, prescribed by specialists, oncology products, or manufacturing technology that is available to only a limited number of firms (Grabowski et al., 2011a). Price and quantity data from IMS Health Inc. were analyzed for 35 conventional (nonbiologic) drugs that experienced generic entry between 1997 and 2003, and those drugs classified as complex were found to have significantly lower levels of generic share and price discounts. Furthermore, complex drugs faced only 2.5 generic entrants 1 year following initial generic entry, whereas noncomplex drugs faced an average of 8.5 generic entrants.
Although data from conventional small-molecule generics should not be directly applied to estimate biosimilar shares following market entry, they suggest that uptake rates for biosimilars may be likely to be significantly lower than those for generics, at least initially. Furthermore, these more complex generic drugs are rated therapeutically equivalent (that is, they have an FDA rating of A) and, therefore, benefit from some automatic substitution. To avoid substitution, physicians need to specify in ‘do not substitute’ orders that prescriptions are to be dispensed as written. At least initially, most biosimilars will not be rated therapeutically equivalent and, therefore, will not be subject to automatic substitution.
Table 3 summarizes other market share and price discount analyses generally based on selective aspects of the US generic market. Most notably, as part of the evaluation of the proposed legislation regarding biosimilars, the Congressional Budget Office (CBO) predicted a penetration rate of 35% with price discounts by biosimilars of 40%. Other estimates of market penetration from a pharmacy benefit management firm, Express Scripts, as well as by Avalere Health, a consulting firm, tend to be somewhat higher than either the Grabowski (2007) or the CBO values, with penetration in the 50–60% range, and somewhat higher discounts in the case of the Avalere study (50% by year 3).
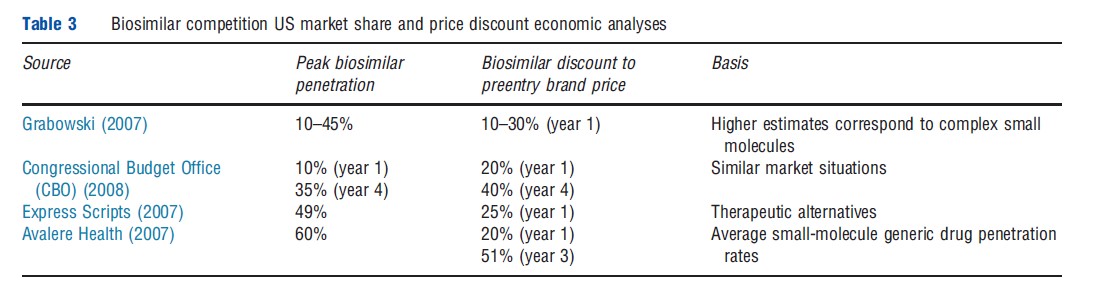
The FDA approval of generic enoxaparin sodium, rated as therapeutically equivalent (having an A-rating) to branded Lovenoxs, provides important data about competitive pricing strategy and market acceptance of generics for a complex, ‘biologic-like’ product. Other notable attributes of Lovenoxs include large expenditures by payers (pregeneric entry sales of more than US$2 billion) and a complicated manufacturing process. Currently, the FDA has approved generic enoxaparin applications from two third-party manufacturers, Sandoz (partnered with Momenta) and Amphastar (partnered with Watson), although the latter is the subject of patent litigation. In addition, there had been for a time an ‘authorized generic’ supplied by Sanofi, the branded manufacturer of Lovenoxs. Sales of generic enoxaparin have been robust and there has been rapid erosion of Lovenoxs’s revenues and market share.
Projected Savings To United States Consumers
The CBO estimated that the provisions in the current health care law establishing a biosimilar pathway would reduce federal budget deficits by US$7 billion over the 2010–2019 period. This finding is consistent with a 2008 CBO study of a similar Senate bill, which estimated a reduction in federal budget deficits of US$6.6 billion and a reduction in biologic drug spending of US$25 billion for the 2009–18 period. Over the full 10-year period, the US$25 billion in reduced biologic drug spending would represent roughly 0.5% of national spending on prescription drugs, valued at wholesale prices. The bulk of these estimated savings accrue in the last 5 years of the 10-year time ranges analyzed. Savings beyond the 10-year period may increase substantially as more biologics lose patent and 12-year exclusivity protections and as scientific advances reduce the cost of developing and producing biosimilars.
A number of the largest-selling biologic products may face losses of some key patent or 12-year exclusivity protections in the coming years. Determining the effective patent-expiry date for any given biologic is fraught with uncertainty because of unknowns such as which patents comprise the portfolio protecting an individual biologic, of which there may be many; the strength of those patents in the face of challenges; and the ability of biosimilar manufacturers to work around existing patents. In November 2011, for example, Amgen announced that it had been issued a patent for the fusion protein etanercept (Enbrels) that could block biosimilar competition until 2028 (the term is 17 years from the date of award, rather than 20 years from the date of application, due to the date of the patent application). Previously, many public sources had anticipated biosimilar entry exposure for Enbrels as early as 2012. Based on a review of patent-expiry information disclosed in manufacturers’ financial reports and supplemented with additional public information from academic literature, research reports, patent filings, and court documents, the earliest publicly reported potential patent-expiry dates for a set of top-selling biologics occur in a timeframe between 2013 and 2018. These biologics include EpogenR/ProcritR, NeulastaR, RemicadeR, RituxanR, and HumiraR (all products having multibillion dollar US sales in 2011). The date when these biologics may actually experience biosimilar market entry under BPCIA depends on many technical, market, regulatory, and legal factors, such as whether entry will be at risk, and the outcome of the patent litigation that is likely to ensue.
The extent of biosimilar cost savings will depend on the timing and number of biosimilar entrants, their market share and price discounts relative to the originator’s product, and the potential competition from the introduction of ‘biobetters’ or next generation products in particular product classes. There is likely to be considerable variation in how competition evolves across biological products reflecting molecule complexity, regulatory criteria, the originating firm’s patent estates, patient populations and physician specialties, as well as changing reimbursement systems and procedures. In contrast to small-molecule generic competition, there is unlikely to be a ‘one-size-fits-all’ pattern for biosimilar competition for the foreseeable future.
Innovation Incentives
As it did with Hatch-Waxman, Congress has attempted to balance the objectives of achieving cost savings from an abbreviated pathway for biosimilars with preserving innovation incentives for new biologics. As discussed earlier, NBEs have been an important source of novel and therapeutically significant medicines. Major advances have occurred for several oncology indications, multiple sclerosis, rheumatoid arthritis, and other life-threatening and disabling illnesses. BPCIA differs from Hatch-Waxman in the term of the data exclusivity period for innovators: BPCIA establishes 12-years data exclusivity period for innovative biologics, whereas Hatch-Waxman establishes a 5-year exclusivity period for NCEs. (The FDA cannot approve an abbreviated application relying on the innovator’s data until these exclusivity periods expire.) Furthermore, as mentioned earlier, the private information exchange process for resolving patent disputes is very different for biologics under the BPCIA than the ‘Orange Book’ public disclosure and Paragraph IV challenge framework for NCEs under Hatch-Waxman.
Regulatory Exclusivity And Patent Protection
The process of discovering and developing a new biologic is a long, costly, and risky venture. DiMasi and Grabowski have estimated that the cost to develop a new FDA-approved biopharmaceutical is US$1.2 billion in risk-adjusted costs, capitalized to 2005 dollars using an 11.5% discount rate (DiMasi and Grabowski, 2007b). DiMasi and Grabowski found that NBEs cost more in the discovery phase, take longer to develop, and require greater capital investment in manufacturing plants than NCEs. They found that the probability of success is higher for biologics than for NCEs, but biologics that fail do so later in the Research and Development (R&D) life cycle. After adjustment for inflation and the different time periods studied, the cost of developing an NBE and an NCE are roughly comparable in value.
Intellectual property protection in the form of patents and regulatory exclusivity are the primary policy instruments by which governments encourages risky investment in R&D for new medicines (together with any tax subsidies or direct financial investment programs that may apply). Regulatory exclusivity and patents have separate but complementary roles. The US government awards patents for inventions based on well-known criteria: novelty, utility, and nonobviousness. A regulatory exclusivity period, however, is needed because after invention a long, risky, and costly R&D process remains for the development of new medicines. Effective patent life is often uncertain because significant patent time elapses before FDA approval and because there is uncertainty associated with the resolution of any patent challenges. As a result, regulatory exclusivity provides a more predictable period of protection. It essentially acts as an ‘insurance policy’ in instances where patents are narrow, uncertain, or near expiry.
The protection afforded by regulatory exclusivity may be particularly important for innovation incentives in biologics to the degree that patents in biologics are narrower in scope than those for small-molecule drugs and more likely to be successfully challenged or circumvented. This may be true to the degree that biologics rely more on process patents, for instance. Given that a biosimilar will be slightly different in its composition and/or manufacturing process, a court may determine that it does not infringe the innovator’s patent. This has the potential to lead to a seemingly contradictory outcome where a biosimilar may be ‘different enough’ not to infringe the innovator’s patents but still ‘similar enough’ to qualify for approval through an abbreviated approval pathway.
As discussed, the BPCIA grants 12 years of exclusivity for innovative biologics during which the FDA may not approve biosimilars referencing them, compared with 5 years of exclusivity for NCEs under the Hatch-Waxman Act, during which an abbrevaited application referencing them cannot be submitted (plus a stay on generic entry for up to 30 months when there is a patent challenge to allow for resolution of litigation). In contrast, the EU has harmonized across member states an ‘8 + 2 + 1’approach for both NCEs and NBEs (consisting of 8 years of data exclusivity, during which generic competitors may not References: the innovator’s data in their applications; 2 years of market exclusivity during which generic marketing authorizations cannot be approved; and a potential additional 1 year of protection for new indications that demonstrate significant clinical benefits over existing therapies that are approved within the first 8 years after the original molecule’s approval).
Economic Analyses Of The 12-Year Exclusivity Period
The US 12-year exclusivity period for innovative biologics was the focus of substantial debate by legislators. The 111th Congress considered bills with exclusivity periods ranging from 5 to 14 years. To provide economic analysis to the legislators, Grabowski (2008) developed a breakeven financial analysis using historical data on R&D costs and revenues for new biologics and the risk-adjusted market return on investment in the industry. Under this model, a representative portfolio of biologic candidates would be expected to ‘break even’ (or recover the average costs of development, manufacturing, promotion, and the industry’s cost of capital) between 12.9 and 16.2 years after launch.
A recently published Monte Carlo simulation model examines the interaction between regulatory exclusivity terms and patent protection periods under different scenarios to highlight the circumstances where each is important in maintaining innovation incentives (Grabowski et al., 2011c). The results of this analysis are generally consistent with Congress’ determination that a regulatory exclusivity period of 12 years appropriately balances objectives for potential cost savings from biosimilar price competition with long-run incentives for investment in innovative biologics. This study finds that when biologic patents are relatively less certain and expected to have shorter effective lifetimes, an exclusivity period of 12 years greatly enhances investment incentives. However, if biologic patents provide relatively strong protection with significant effective patent life remaining at approval, patents alone will be sufficient to maintain investment incentives in most cases. In those instances, however, the 12-year exclusivity period has only a minimal effect on the timing of potential biosimilar entry and consequently on healthcare costs.
It remains unclear whether the longer exclusivity periods for biologics compared with chemical entities will tilt R&D incentives toward large molecules and whether Congress will consider harmonizing these periods, as is currently the case in the EU.
The Resolution Of Patent Challenges
Hatch-Waxman also featured Paragraph IV 180-day exclusivity provisions, under which generic manufacturers could challenge the legitimacy of branded manufacturers’ patents or claim that generic entry would not infringe them. Over time, as the law and economic benefits to generics were established, the likelihood of Paragraph IV challenges increased and most drugs became subject to challenges (Berndt et al., 2007; Grabowski et al., 2011a). This has led to uncertainty regarding the effective patent term for new drug introductions, as well as substantial litigation costs early in the product life cycle.
Under the BPCIA, an abbreviated application for a biosimilar can be filed after 4 years. The filing of an application triggers a series of potentially complex private information exchanges between the biosimilar applicant and References: product innovator. These exchanges are followed by negotiations and a process for instituting litigation on the core patents, when necessary. Congress has crafted these patent provisions while eliminating the incentive for litigation associated with a 180-day exclusivity period for the first filer in a successful challenge, as well as the automatic 30-month stay on entry under Hatch-Waxman. By instituting this potentially complex structured process for biologics, legislators hoped that patent disputes would be resolved before the expiration of the 12-year exclusivity period so that biosimilars can enter in a timely fashion. Generic manufacturers have raised concerns about the need to divulge proprietary information, and whether these rules will achieve their intended effects remains unknown.
Firms pursuing a biosimilar strategy could also choose to file a full BLA rather than an abbreviated application. Under the patent resolution provisions of the BPCIA, firms filing an abbreviated biosimilar application are required to disclose information about their manufacturing process and identify potential patent conflicts. By choosing instead to file a full BLA, the biosimilar firm would avoid this disclosure requirement, and, if approved, also be able to enter before the expiration of the 12-year exclusivity period. However, the firm needs to weigh these benefits against the additional investment of expenditures and time associated with filing a full BLA for a biosimilar product. Several firms apparently are considering this strategic option. Teva recently relied on a full BLA filing for its G-CSF filgrastim product, although the original submission to the FDA predated the establishment of a biosimilar pathway in the US. In Europe, the same Teva product is marketed under the name TevagrastimR and was approved through an abbreviated biosimilar application for the reference product Neupogen (Table 1). The product is scheduled to be launched in the US in late 2013 under a patent settlement with Amgen.
Summary And Conclusion
Biologics have accounted for a significant number of innovative medicines over the past three decades. At the same time, they account for a growing share of drug expenditures in some countries. Policymakers have anticipated the introduction of biosimilars mitigating these cost pressures. Biosimilars have been introduced in various EU countries beginning in 2007. The extent of biosimilar penetration for the biological entities, erythropoietin, G-CSF, and somatropin has varied substantially across therapies within a country and across countries for the same therapy. Germany has experienced the greatest initial uptake of biosimilars reflecting targeted incentives quotas and related factors.
The new US law is designed to balance the objectives of achieving cost savings in the current period and preserving incentives for continued innovation in the future. A number of leading biologic products with significant sales in the US are expected to experience some patent expiration in the next decade, so cost savings could grow significantly over time, depending on how other factors such as regulation, reimbursement, and intellectual property litigation evolve over this period.
In terms of maintaining incentives for future innovation, the US law provides for a 12-year exclusivity period after an innovator’s product is approved before a biosimilar referencing can be approved utilizing an abbreviated pathway. This 12-year exclusivity period provides an important ‘insurance policy’ to the patent system and could be important in the case of biologics where patents may prove to provide less certain protection than those for NCEs. Analysis of a portfolio of representative biological products indicates that 12 years or more of exclusivity from patents or regulatory provisions is generally consistent with achieving breakeven returns that provide a risk-adjusted return on capital and R&D investments.
A number of important issues remain for future research, including how the new law will affect industry structure and incentives for undertaking R&D for biologics versus NCEs. As was the case with Hatch-Waxman, change may be gradual at first, but over time the new law could lead to profound changes in the economics and organization of the biopharmaceutical industry.
References:
- Avalere Health (2007). Modeling federal cost savings from follow-on biologics (study author King, R.).
- Berndt, E., Mortimer, R., Bhattacharjya, A., Parece, A. and Tuttle, E. (2007). Authorized generic drugs, price competition, and consumers’ welfare. Health Affairs 790, 792–797.
- Chauhan, D., Towse, A. and Mestre-Ferrandiz, J. (2008). The market for biosimilars: evolution and policy options. Office of Health and Economics Briefing, No. 45, 12–14.
- Congressional Budget Office (CBO) (2008). S.1695, Biologics Price Competition and Innovation Act of 2007.
- DiMasi, J. and Grabowski, H. (2007a). The economics of new oncology drug development. Journal of Clinical Oncology 209, 214–215.
- DiMasi, J. and Grabowski, H. (2007b). The cost of biopharmaceutical R&D: Is biotech different? Managerial & Decision Economics 469–475.
- Express Scripts (2007). Potential savings of biogenerics in the United States (study authors Miller, S. and Houts, J.).
- Food and Drug Administration (FDA) (2012a). Scientific considerations in demonstrating biosimilarity to a References: product.
- Food and Drug Administration (FDA) (2012b). Biosimilars: Questions and answers regarding implementation of the Biologics Price Competition and Innovation Act of 2009.
- Federal Trade Commission (2002). Generic drug entry prior to patent expiration: An FTC study.
- Grabowski, H. (2007). Competition between generic and branded drugs. In Sloan, F. A. and Hsieh, C-R. (eds.) Pharmaceutical innovation: Incentives, competition, and cost–benefit analysis in international perspective, pp. 153–173. New York, NY: Cambridge University Press.
- Grabowski, H. (2008). Follow-on biologics: Data exclusivity and the balance between innovation and competition. Nature Reviews Drug Discovery 479, 479–487.
- Grabowski, H., Cockburn, I. and Long, G. (2006). The market for follow-on biologics: How will it evolve? Health Affairs 1291, 1291–1301.
- Grabowski, H., Kyle, M., Mortimer, R., Long, G. and Kirson, N. (2011a). Evolving brand-name and generic drug competition may warrant a revision of the Hatch-Waxman Act. Health Affairs 30, 2157–2166.
- Grabowski, H., Lewis, T., Guha, R., et al. (2012). Does generic entry always increase consumer welfare? Food and Drug Law Journal.
- Grabowski, H., Long, G. and Mortimer, R. (2011c). Data exclusivity for biologics. Nature Reviews Drug Discovery 15, 15–16.
- Grabowski, H., Ridley, D. and Schulman, K. (2007). Entry and competition in generic biologics. Managerial & Decision Economics 28(4–5), 439–447.
- Grabowski, H. and Wang, R. (2006). The quantity and quality of worldwide new drug introductions, 1982–2003. Health Affairs 25(2), 452–460.
- Hargrave, E., Hoadley, J., Merrell, K. (2010). Medicare Part D Formularies, 2006–2010: A Chartbook, Report to the Medicare Payment Advisory Commission.
- Heldman, P. (2008). Follow-on biologic market: Initial lessons and challenges ahead. Potomac Research Group, Presentation to the Federal Trade Commission.
- IMS (2011a). Shaping the biosimilars opportunity: A global perspective on the evolving biosimilars landscape.
- IMS (2011b). The Use of Medicines in the United States: Review of 2010. IMS Institute for Healthcare Informatics.
- Kambhammettu, S. (2008). The European biosimilars market: Trends and key success factors. Scicast Special Reports.
- Kozlowski, S., Woodcock, J., Midthun, K. and Behrman, S. R. (2011). Developing the nation’s biosimilar program. New England Journal of Medicine 364(5), 385–388.
- Smith, I., Procter, M., Gelber, R. D., et al. (2007). 2-Year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: A randomized controlled trial. Lancet 369(9555), 29–36.
- Sokolovsky, J., Miller, H. (2009). Medicare payment systems and follow-on biologics, Medicare Payment Advisory Commission.
- Trusheim, M. R., Aitken, M. L. and Berndt, E. R. (2010). Characterizing markets for biopharmaceutical innovations: do biologics differ from molecules? Forum for Health Economics & Policy (Frontiers in Health Policy Research) 13(1), 1–45. The Berkeley Electronic Press.
- Weaver, A. L. (2004). The impact of new biologicals in the treatment of rheumatoid arthritis. Rheumatology 43(Supplement 3), iii17–iii23.
- Grabowski, H., Long, G. and Mortimer, R. (2011b). Implementation of the biosimilar pathway: Economic and policy issues. Seton Hall Law Review 41(2), 511–557.
- Rovira, J., Espin, J., Garcia, L., and de Labry, A. O. (2011). The impact of biosimilars entry in EU markets. Andalusian School of Public Health.